Introduction
In genetic association analysis, allele coding discordance is one commonly encountered problem that many researchers struggle with, especially when two different groups compare results or perform meta-analysis between cohorts. This issue arise between different people and different computer algorithms tend to use different allele coding schemes: some use ACGT alleles based on forward strand of the human genome assembly, some use Illumina's TOP alleles, and some use Illumina's AB alleles. This software is written to convert the allele coding shemes so that results from different groups are comparable to each other. The input file for this program is a BIM file that was generated by the PLINK program, since the file has a simple format and can be easily manipulated. If the user has a PED file instead, it's easy to convert it to BIM file using PLINK, then convert back to PED file.
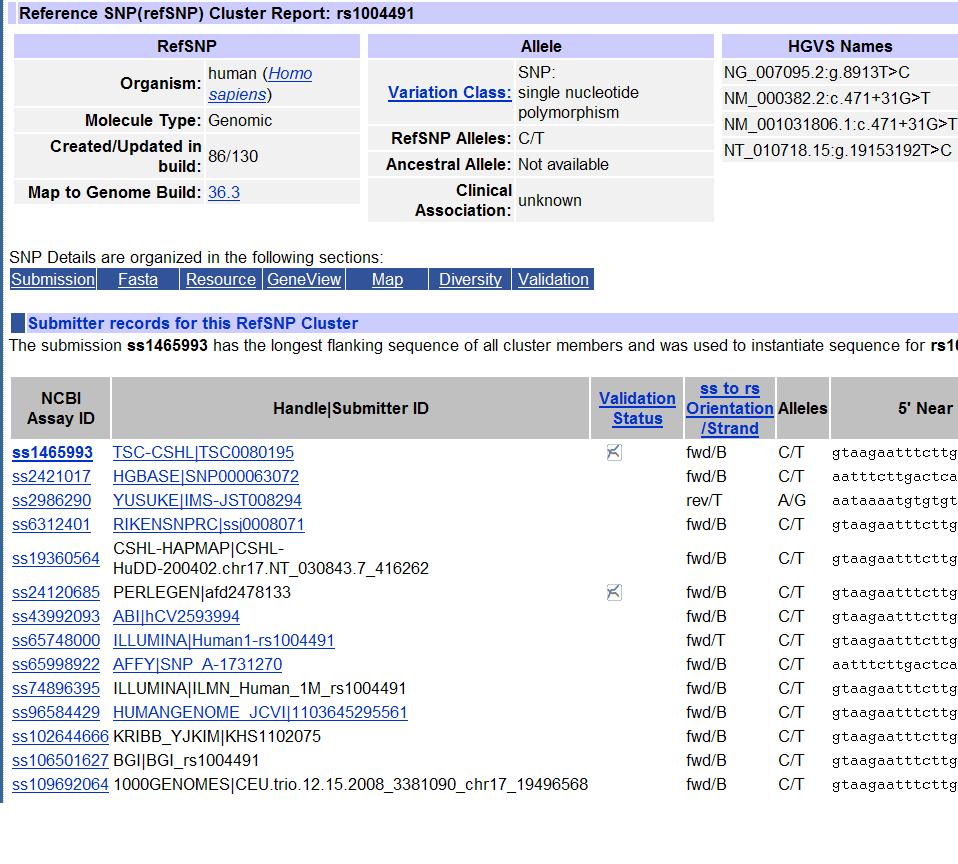
Below I briefly describe several commonly used allele coding schemes. To make things easier to understand, let's first look at a screen shot from dbSNP. Simply search "rs1004491" in dbSNP, and click the search result, and this screen will show up. It tells that the SNP rs1004491 is compiled based on about a dozen individual submissions (with ss identifier) from various sites. The SNP is mapped to NCBI build 36, with refSNP allele as C/T in dbSNP, which is in forward strand. You will also notice that while the vast majority of sources indicate that the forward strand is equal to BOTTOM strand, one source (from Illumina Human1 array) indicate otherwise so it is likely to be a wrong annotation from Illumina (even though Illumina was the inventor of the TOP/BOTTOM concept). These types of mistakes happen a lot in practice.
- forward allele
This is the allele that most people should be working on. It refers to the allele in the forward strand in a reference genome. However, different versions of reference genomes tend to differ in specific locations with common SNPs; for example, if GRCh37 has a minor allele in a SNP in the reference strand, GRCh38 tends to change this allele back to major allele (I have seen many examples like this already), so forward allele ideally should always be coupled with the genome build to uniquely identify a SNP. Unfortunately most people only say "forward strand" and they never accompany this statement with a genome build.
Note that in April 2017 I modified this article to add explanation for forward allele. The GenGen software in GitHub itself was working correctly on forward alleles in the past a few years, but I never updated the documentation so it may have caused some confusion among users. So I emphasize here again: forward allele does not equate dbSNP allele! A -strandfile is necessary to ensure that forward allele is computed correctly from dbSNP allele.
- dbSNP allele
This is a commonly used allele coding scheme, although it has many intrinsic problems that I'll describe below. In most cases it is identical as forward strand allele, but there are some exceptions, so users need to exercise caution. The scheme assumes that a "forward strand" for a given genome is already known, and that the two alternative alleles are defined with respect to the forward strand (as opposed to the reverse strand). For example, suppose that a SNP has C allele in the forward strand in reference genome sequence (and has a G allele in the reverse strand); an alternative sequence is found in the population that has A allele in the reverse strand (and hence T allele in the forward strand), then the SNP is then coded with C and T alleles. In the above example, you can see that this SNP is a C/T SNP based on forward strand (or a A/G SNP based on reverse strand).
One obvious problem with this coding scheme is that one must know the "forward strand" with certainty, but this is usually not the case. Even for the well studied human genome, there are still many holes to be filled. It is possible that a sequence is assembled into the forward strand in 2004 human genome assembly, but it becomes reverse strand in the 2006 human genome assembly. It is also possible that a sequence cannot be assigned into either forward or reverse strand, so it's annotated as something like chr1_random or a random contig in many genome databases. It is also possible that a stretch of forward strand becomes reverse strand in a particular subject due to inversion in this region. These types of situations becomes much worse for other organisms that have only low fold coverage genome sequences: it's simply not feasible to assign many contigs correctly into either a forward or a reverse strand, so even if a SNP is known based on sequencing many subjects, the correct "forward strand" coding cannot be inferred confidently.
To ensure the correct identification of forward strand for dbSNP records, the users need to obtain a strandfile, which can be compiled from say a snp147 file in UCSC Genome Browser (or if you use ANNOVAR, just do a -downdb snp147 should work) with a cut command to cut the appropriate columns in the file.
- Illumina A/B allele, 1/2 allele, and TOP allele
Illumina's A/B allele coding, or TOP/BOT strand definition, is explained in here in detail by Illumina. The allele coding method solves the problem aformentioned, that is, the alleles are not dependent on the specific genome assembly, but are based on the actual polymorphism itself. Briefly, if one of the two polymorphism is A or T, and the other one is C or G, then the A or T is refered to as A allele, and the C or G is refered to as B allele, and the strand with A or T is refered to as TOP and BOT strand, respectively. If the polymorphism is A/T or C/G, then walk through the surrouding sequence (the two nucleotides up or downstream of the SNP) to find a pair of unambiguous nucleotides, and then a similar rule is applied: if A or T is on 5' side of the SNP, then it's a TOP strand otherwise it's a BOT strand. For TOP strand, A and B allele denote A and T (or C and G), respectively; whereas for BOT strand, A and B allele denote T and A (or G and C), respectively. The Illumina's coding scheme does not rely on definition of a forward strand (hence correct genome assembly), so it almost always ensure consistency between genome builds and ensures instant allele designation for newly sequenced genomic sequences or unassembled genomic sequences. As the above figure shows, dbSNP has already adopted the Illumina's coding scheme in their annotations: fwd/B means that "forward" strand of dbSNP corresponds to "BOT" strand of Illumina's coding scheme. Also note that there is a "fwd/T" in the figure above: it clearly indicates an annotation error: such errors are indeed quite prevalent! Sometimes, people often use 1/2 to denote Illumina's A/B allele, since numeric coding is more convenient in many scenarios and since some old association software only recognize numeric coded alleles.
When exporting genotypes from the Illumina BeadStudio software, the user can choose AB genotypes, or ACGT genotypes (commonly refered to as "TOP alleles"), or forward strand genotype in newer version of the software. The TOP alleles is the allele on the TOP strand, which may or may not be the forward strand: see the example above, the "fwd/B" means that dbSNP's forward strand corresponds to Illumina's BOT strand, so the "TOP allele" is the opposite as the "forward strand allele". Unfortunately many users simply do not know or understand what is "TOP allele": they simply take for granted that "TOP" means "forward" and then complain that there are many discordances when merging two different data sets (one coded as forward strand and one exported from BeadStudio). The convert_bim_allele.pl program that I describe in this article will solve problems like this.
- Affymetrix A/B allele
Note that Affymetrix also has a A/B allele designation but as far as I can tell, nobody actually uses it in any circumstance except when doing CNV analysis (for example, in PennCNV-Affy, the A/B alleles are explicitly used to relate frequency information of B allele from external databases to the observed B alleles). Virtually all SNP genotype calling algorithms for the Affymetrix platform generate actual ACGT allele calls in forward strand. Unfortunately, annotating alleles in forward strand is a difficult task, and there are many annotation errors in the library file provided by Affymetrix, or those produced by any third-party software. The user will have to exercise caution when merging genotyping calls from different software together, when using the Affymetrix platform. For example, when merging two data sets on the same platform but on different subjects, it's always a good idea to calculate allele frequencies for all SNPs in these two data sets and compare them to ensure consistent frequency spectrums.
- HapMap data set
Unfortunately, many errors exist in HapMap "foward strand" haplotype data as well, and given that genotype imputation has become very popular these days, it is important to recognize this fact and handle it appropriately. For Illumina Infinium assays, since no A/T and C/G SNP exists, typically this is not a big problem and an "autoflip" function can be used in imputation software to handle the allele-flipping issues. For Illumina GodenGate assays and for Affymetrix assays, this is a practical issue, since one will have to change "correct" genotype calls (in forward strand) into incorrect ones simply to accormodate the erraneous HapMap data for the purpose of imputation. Essentially, the user will have to do extra work when merging imputated genotypes from HapMap with another genotype data set, or when performing meta-analysis involving both imputated data and genotyped data. Hopefully, the convert_bim_allele.pl program may make this easier.
- PLINK 1/2 allele
Some association or linkage software requires that alleles be coded as nuemric values, therefore one may want to convert ACGT alleles into 1/2 alleles. Softwares such as PLINK can perform this task, using --recode12 argument. However, the 1/2 coding by PLINK cannot be decoded back to ACGT, since 1 and 2 denote minor/major allele, respectively, and these annotations differ between data sets. When getting a genotype data file with 1/2 allele coding from other people, it is always necessary to ask whether the alleles are coded as Illumina's A/B alleles or simply recoded using PLINK; if it's the latter, it is best to ask for original genotype data before doing anything with the data.
Input files
The program requires two main input files, an PLINK-formatted BIM file, a SNPTable file mapping different allele coding schemes. Their formats are briefly described below.
- BIM file
The BIM file can be generated by the PLINK software using the --make-bed argument, see details here.
An example file is shown below:
[kai@beta ~/project/]$ head 45snp.bim
1 rs2269241 0 63820792 C T
1 rs6679677 0 114015850 A C
1 rs1323296 0 189269165 A G
1 rs3024505 0 203328299 A G
2 rs10175906 0 12610746 G C
2 rs2041756 0 102508428 A G
2 rs7608315 0 163039230 C A
2 rs3087243 0 204564425 A G
3 rs6441961 0 46327388 T C
4 rs17698094 0 25782567 G A
Each line contains information for one marker with four fields, including chromosome, marker name, genetic position, physical position, and two alleles (when PLINK makes the file, the minor allele is always listed before major allele, but obviously this arrangments may differ between different data sets).
- SNPTable file
The SNPTable file is a tab-delimited file that annotates the allele designation for each SNP. For Illumina arrays, one can easily generate such a file from the BeadStudio software: simply click the "SNPTable" tab after opening a project file, then use "column chooser" to select the "Name", "SNP", "ILMN Strand" and "Customer Strand" columns only, then click "export" to save the output to a file with the four columns. In the GenGen software package, I have provided a SNPTable file for HumanHap550&610 arrays, as well as a file for HumanHap1M v1 and v3 arrays. Annotation file for other arrays can be generated by the user using the above procedure.
To explain this in more detail, see the first 10 lines of a SNPTable file below:
[kai@beta ~/project/ head hh550_610.snptable
Name SNP ILMN Strand Customer Strand
200003 [T/C] BOT TOP
200006 [A/G] TOP BOT
200047 [T/C] BOT TOP
200050 [C/G] TOP BOT
200052 [T/A] BOT TOP
200053 [T/C] BOT BOT
200070 [G/C] BOT BOT
200078 [C/G] TOP BOT
200087 [T/G] BOT BOT
The first line is a header line that explains the meaning of each column, and each subsequent columns annotate one SNP (the SNPs shown above do not have rs identifiers, but they are indeed SNPs in the Illumina array).
For arrays with indels, the alleles are shown as D (deletion) and I (insertion), rather than ACGT. For example, see below:
[kai@beta ~/project/]$ fgrep D ~/project/lib/hh1m.snptable | head
rs17838100 [D/I] P P
rs3093288 [I/D] M M
rs8177128 [D/I] P P
rs4251989 [I/D] M M
rs11572837 [I/D] M M
rs2307321 [D/I] P P
rs3138160 [I/D] M M
rs28921973 [D/I] M M
rs17223353 [D/I] P P
rs4647073 [D/I] M M
Several arguments can control the behavior of the convert_bim_allele.pl program for handling D/I alleles.
Convert allele coding schemes
Four different allele coding types are supported by the convert_bim_allele.pl program: ilmn12, ilmnab, dbsnp, forward and top. The ilmn12 and ilmnab refers to Illumina's A/B coding, dbSNP refers to forward strand coding, and top refers to the TOP strand coding. The program requires an --intype and an --outtype arguments, as well as an --outfile argument to store the new BIM files.
[kai@beta ~]$ convert_bim_allele.pl
Usage:
convert_bim_allele.pl [arguments] <bimfile> <snptablefile>
Optional arguments:
-h, --help print help message
-m, --man print complete documentation
-v, --verbose use verbose output
--intype <ilmn12|ilmnab|top|dbsnp|forward> specify input type
--outtype <ilmn12|ilmnab|top|dbsnp|forward> specify output type
--force force execution when allele mismatch is detected (output zero allele)
--replacezero replace the zero (unobserved) allele by known allele (default: output zero allele)
--outfile <file> specify output file name (default: STDOUT)
--autoflip automatically flip strand where appropriate
--strandfile <file> specify a file with dbSNP strand information
Function: convert a BIM file so that the allele names are changed to the
specified outtype.
Notes: ilmn12=Illumina AB allele designation coded as 1 and 2
ilmnab=Illumina AB allele designation
top =Illumina Top Allele call
dbsnp =dbSNP (not necessarily forward) allele designation
forward=forward strand in reference genome
Example: convert_bim_allele.pl old.bim hh550_610.snptable -outfile new.bim -intype ilmnab -outtype top
convert_bim_allele.pl old.bim hh550_610.snptable -outfile new.bim -intype top -outtype dbsnp
- Convert Illumina A/B (or 1/2) allele coding to other coding types
Given a BIM file with Illumina's A/B coding (or 1/2 coding, where 1 denotes A and 2 denotes B), one can run the simple commands below to convert the allele coding scheme in the file.
[kai@beta ~]$ cat test.bim
1 rs2476601 0 114179091 A B
2 rs3747517 0 162837070 A B
2 rs231804 0 204416891 B A
4 rs7699742 0 123483935 B A
6 rs3129871 0 32514320 A B
6 rs3757247 0 91014184 A B
10 rs12251307 0 6163501 A B
11 rs4244808 0 2119686 B A
12 rs1701704 0 54698754 B A
12 rs17696736 0 110971201 B A
15 rs2870085 0 77021323 A B
16 rs725613 0 11077184 B A
18 rs1893217 0 12799340 B A
22 rs229527 0 35911431 A B
[kai@beta ~]$ convert_bim_allele.pl test.bim ~/project/lib/hh550_610.snptable -intype ilmnab -outtype top -outfile test.top.bim
NOTICE: Reading SNP Table file /home/kai/project/lib/hh550_610.snptable ... Done with 640728 SNPs
NOTICE: The new bim file will be written to test.top.bim ... Done
[kai@beta ~]$ cat test.top.bim
1 rs2476601 0 114179091 A G
2 rs3747517 0 162837070 A G
2 rs231804 0 204416891 G A
4 rs7699742 0 123483935 G A
6 rs3129871 0 32514320 A C
6 rs3757247 0 91014184 A G
10 rs12251307 0 6163501 A G
11 rs4244808 0 2119686 C A
12 rs1701704 0 54698754 C A
12 rs17696736 0 110971201 G A
15 rs2870085 0 77021323 A G
16 rs725613 0 11077184 C A
18 rs1893217 0 12799340 G A
22 rs229527 0 35911431 A C
The above example convert the Illumina A/B coding into Illumina's TOP strand coding. Note that these SNPs are all A/C or A/G SNPs in the TOP strand coding, and there is no T allele at all.
[kai@beta ~]$ convert_bim_allele.pl test.bim ~/project/lib/hh550_610.snptable -intype ilmnab -outtype dbsnp -outfile test.dbsnp.bim
NOTICE: Reading SNP Table file /home/kai/project/lib/hh550_610.snptable ... Done with 640728 SNPs
NOTICE: The new bim file will be written to test.dbsnp.bim ... Done
[kai@beta ~]$ cat test.dbsnp.bim
1 rs2476601 0 114179091 A G
2 rs3747517 0 162837070 A G
2 rs231804 0 204416891 C T
4 rs7699742 0 123483935 C T
6 rs3129871 0 32514320 A C
6 rs3757247 0 91014184 A G
10 rs12251307 0 6163501 T C
11 rs4244808 0 2119686 C A
12 rs1701704 0 54698754 C A
12 rs17696736 0 110971201 G A
15 rs2870085 0 77021323 T C
16 rs725613 0 11077184 C A
18 rs1893217 0 12799340 C T
22 rs229527 0 35911431 T G
The above example convert the Illumina's A/B coding into dbSNP forward strand coding. Note that unlike TOP strand coding, some SNPs are C/T or T/G SNPs. Since the file is generated from an Infinium assay, there is no A/T or C/G SNPs; if the genotype data is from a GoldenGate assay or from Affymetrix array, there will be A/T or C/G SNPs.
- Convert Illumina's TOP allele coding to other coding types
An example is given below. The --intype of top is used.
[kai@beta ~]$ convert_bim_allele.pl test.top.bim ~/project/lib/hh550_610.snptable -intype top -outtype dbsnp -outfile test.dbsnp.bim
NOTICE: Reading SNP Table file /home/kai/project/lib/hh550_610.snptable ... Done with 640728 SNPs
NOTICE: The new bim file will be written to test.dbsnp.bim ... Done
[kai@beta ~]$ cat test.dbsnp.bim
1 rs2476601 0 114179091 A G
2 rs3747517 0 162837070 A G
2 rs231804 0 204416891 C T
4 rs7699742 0 123483935 C T
6 rs3129871 0 32514320 A C
6 rs3757247 0 91014184 A G
10 rs12251307 0 6163501 T C
11 rs4244808 0 2119686 C A
12 rs1701704 0 54698754 C A
12 rs17696736 0 110971201 G A
15 rs2870085 0 77021323 T C
16 rs725613 0 11077184 C A
18 rs1893217 0 12799340 C T
22 rs229527 0 35911431 T G
- Convert dbSNP strand coding to other coding types
An example is given below. The --intype of dbsnp is used, which denotes that the alleles are in forward strand orientation.
[kai@beta ~]$ convert_bim_allele.pl test.dbsnp.bim ~/project/lib/hh550_610.snptable -intype dbsnp -outtype ilmn12 -outfile test.ilmn12.bim
NOTICE: Reading SNP Table file /home/kai/project/lib/hh550_610.snptable ... Done with 640728 SNPs
NOTICE: The new bim file will be written to test.ilmn12.bim ... Done
[kai@beta ~]$ cat test.ilmn12.bim
1 rs2476601 0 114179091 1 2
2 rs3747517 0 162837070 1 2
2 rs231804 0 204416891 2 1
4 rs7699742 0 123483935 2 1
6 rs3129871 0 32514320 1 2
6 rs3757247 0 91014184 1 2
10 rs12251307 0 6163501 1 2
11 rs4244808 0 2119686 2 1
12 rs1701704 0 54698754 2 1
12 rs17696736 0 110971201 2 1
15 rs2870085 0 77021323 1 2
16 rs725613 0 11077184 2 1
18 rs1893217 0 12799340 2 1
22 rs229527 0 35911431 1 2
Advanced topics
- Handling missing alleles in BIM file
Sometimes the BIM file contains only one allele for a SNP, since the other allele is never observed in genotype data. The missing allele is shown as "0" in the BIM file (fourth column, since it denotes minor allele). For example, the corresponding line in the BIM file might be "2 rs231804 0 204416891 0 A ", indicating that only allele T is observed in the genotype data.
If --fillzero argument is set , the missing allele will be filled. For example, the aformentioned example will become "2 rs231804 0 204416891 C T". If both alleles for a SNP is missing, then the two missing alleles are both filled in random order.
- Handling insertions and deletions
Some SNPs are indels, that is, they represent the insertion or deletion of one or multiple nucleotides, so the alleles cannot be denoted by ACGT. The alleles for the indels can be denoted by D and I in the BIM files, since they are used in the SNPTable file for Illumina arrays. If the BIM file (in dbsnp or top coding) use d/i or something else to denote indels, before conversion, it is important to change them to D/I coding so that the program does not complain about error.